Cyclospora cayetanensis is not an easy pathogen to deal with. It cannot be grown in the lab. It cannot be studied in lab animals. It cannot be inactivated by the chemical treatments that kill bacteria.
People become infected by eating contaminated food or drinking contaminated water. The pathogen takes up its home in the lining of the intestines, multiplies, and is shed in an inactive form (oocysts) in feces. Human feces. Over a period of days, the oocysts self-fertilize through asexual or sexual reproduction and become embryos capable of infecting their next human victim.
Cyclospora does not multiply outside of a host animal. In the case of C. cayetanensis, that host—the only host—is the human body.
For a field crop such as iceberg lettuce to be contaminated with Cyclospora, it must have come into direct contact with human feces contained either in irrigation water or in fertilizer or soil amendments.
CDC’s slips are showing
The CDC’s reporting of both the Taylor Farms lettuce outbreak and of the country-wide surge in Cyclospora reports has been slipshod and unclear.
When the CDC posts a new outbreak investigation notice, the information usually includes both a map showing the distribution of outbreak cases by state and what is called an Epi Curve—a chart that tracks the case reports by date of illness onset. Both of those visual aides are accompanied by data tables.
The CDC posted its initial notice of a Cyclospora outbreak on July 14, 2026. The notice indicated the number of cases confirmed to be part of the outbreak, and the four states where cases had been confirmed, but did not offer either a breakdown of cases by state or an Epi Curve. Instead, the notice simply advised that the earliest outbreak victims reported becoming ill on June 22, 2026.
In its July 17th update, the CDC added a case distribution map, updated the number of states to five from four, increased the number of confirmed cases to 1,644, and modified the earliest illness onset date to May 13, 2026. There was still no Epi Curve included with the update.
It was July 24th before the CDC finally added an Epi Curve to its outbreak investigation notice. They also updated the number of confirmed cases to 1,947 and added four more states to the outbreak case map. The July 24th notice also modified the illness onset date range once again, advancing the earliest illness onset from May 13th to June 22nd (tip of the keyboard to Bill Marler for pointing out this date discrepancy).
In doing so, the CDC removed at least 210 cases from the tally (Michigan dropped from 1,141 to 931). We don’t know how many more cases were removed, as we do not have an Epi Curve that shows the number of cases by onset date between May 13th and June 22nd.
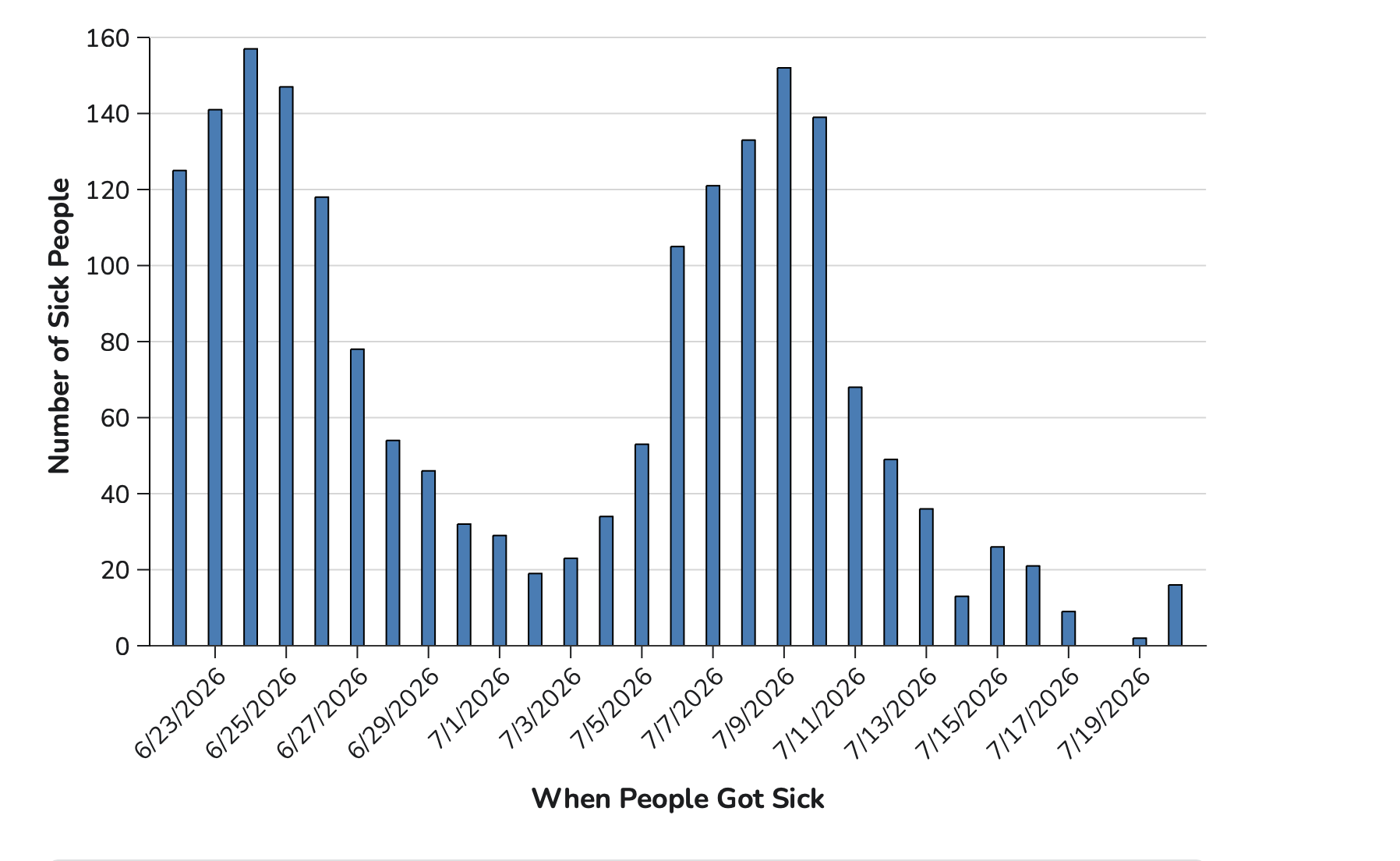
Unusual Epi Curve
This is the Epi Curve that the CDC posted on July 24th. Notice two things: (1) the curve begins right near a peak in case numbers and (2) there are twin peaks.
For comparison, here are two Epi Curves from previous Cyclospora outbreaks, one from 2019 and the other from 2020
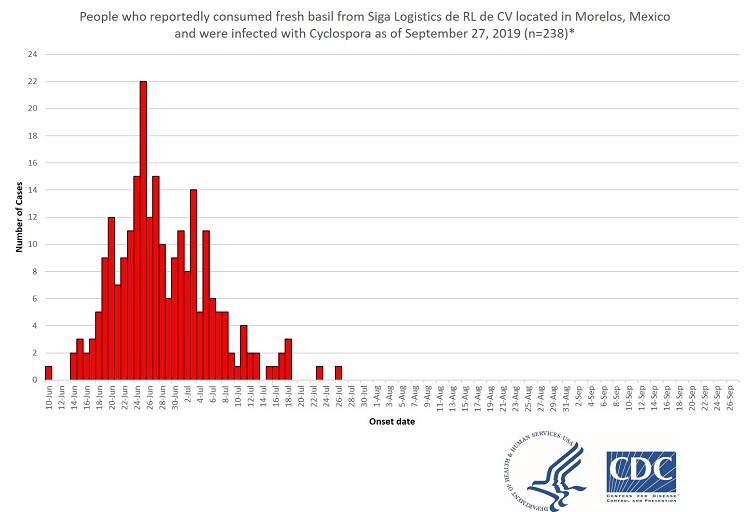
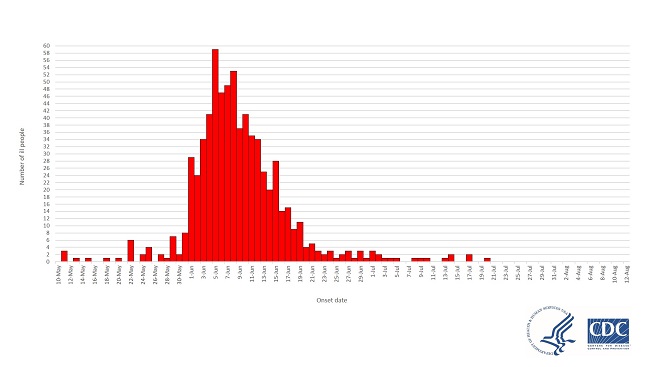
Notice the difference? In both of these older Cyclospora outbreaks associated with leafy greans, there was a build-up of cases. Just a few, at first, then an acceleration to a peak followed by a drop-off.
An outbreak does not burst onto the scene at the peak of its strength.
The twin peaks of the current outbreak also are very unusual. This could be a reporting glitch, with some states lagging in their reports to the CDC. It could be a data collation and analysis lag at the CDC. Or it might be real. We’ll have to wait and see.
Cyclospora surveillance data don’t compute
On July 24th, the CDC also updated its Cyclospora surveillance data, reporting 4,173 laboratory-confirmed domestic cases across 41 states, with an additional 7,400+ cases that are not yet lab-confirmed.
Setting aside the reporting and data-processing lags that have resulted in states posting higher numbers on their own websites than those revealed in the CDC report, there are discrepancies between the data in the surveillance table and the data reported for the Taylor Farms outbreak.
Specifically, the CDC reports a total of “301 to 500” Ohio cases in the surveillance table, but 639 Ohio outbreak cases. Similarly, West Virginia is shown as having reported 42 outbreak cases; yet, only “11 to 30” total cases in the surveillance table.
And while we’re at it, since when does the CDC indicate case-count data by range rather than by actual numbers of cases? The agency knows very well what the case counts are by state. It uses those counts to arrive at the national total. Under normal circumstances, the case-count table shows those individual numbers and NOT a relatively meaningless range, such as the “901 to 2100” cases attributed to Michigan or the “301 to 500” cases shown for Ohio.
What is going on here?
Important surveillance tool tossed in the trash

Once upon a time, the CDC actively searched for incipient Cyclospora infections. The FoodNet program, which covers ten states plus some counties in California, provides active surveillance of the incidence of foodborne diseases in about 16% of the population. In the words of the CDC, “FoodNet is an active surveillance system, meaning that public health officials routinely communicate with more than 700 clinical laboratories serving the surveillance area to identify new infections and conduct periodic audits to ensure that all infections are reported.” (emphasis added).
While FoodNet was not designed specifically to detect foodborne disease outbreaks, it was an important tool to ensure that the systems the CDC relies upon for outbreak detection are functioning as they were meant to.
On July 1, 2025, the CDC changed the reporting requirement for Cyclospora and several other foodborne pathogens from mandatory to optional. Only Salmonella and shiga toxin-producing E. coli remain from the original ten pathogens or conditions that formed the basis for the original FoodNet program.
The FDA’s unforced error
On July 18, 2026, the FDA announced that it had found Cyclospora in a sample of Taylor Farms de Mexico’s shredded iceberg lettuce. One day later, the agency retracted that announcement, explaining that, “Due to the complexity in detection of Cyclospora, FDA laboratory experts re-reviewed the sample results and have concluded that the finding does not represent true amplification and should be considered a false positive.”
What a rookie error!
The sample result should have been reviewed by the agency’s experts before the result was announced, not afterwards. This fumbling of the ball significantly damaged the credibility of the rest of the evidence supporting the FDA’s conclusion that Taylor Farms de Mexico was the source of the outbreak. Instead, it spawned a rash of conspiracy theories on social media. Suggestions that Taylor Farms had twisted arms to get the FDA to retract its lab findings. Hints of payoffs at the most senior levels of the government.
Also feeding those conspiracy theories was the FDA’s apparent reluctance to name Taylor Farms de Mexico as the source of the contaminated lettuce, focussing instead on Taco Bell, the franchise chain named by most of the interviewed outbreak patients as the place where they had consumed iceberg lettuce.
On July 16th, the FDA said that it had identified “…a single supplier of iceberg lettuce from Mexico used by Taco Bell locations where sick people ate before becoming ill,” and was “…working directly with the identified supplier to determine if potentially contaminated shredded iceberg lettuce remains on the market.” It was only on July 17th, the day Taylor Farms de Mexico announced a product recall, that the FDA added the company’s name to the information in the outbreak investigation report on the agency’s website.
What is worse than decimation?
The term “decimation” derives from the old Roman custom of disciplining a military legion by executing 10% of the ranks.
What has taken place within the CDC and the FDA under Trump 2.0 is far worse. The cuts initiated in 2025 by Elon Musk’s so-called “Department of Government Efficiency” were catastrophic. Senior employees who had been promoted into new jobs within the prior two years were ‘probationary’ and therefore able to be terminated with minimal red tape. Similarly, the enthusiastic young blood at the CDC, the interns who had been hired to train as disease detectives, were easy targets for dismissal.
To add to the confusion, the entire communications staff at both agencies was laid off (although some were later ‘invited’ back), and it shows.
It shows in the reduced frequency of outbreak investigation updates on the CDC website.
It shows in the poorly written recall notices on the FDA website.
The old FDA—the agency I once knew and respected—would not have made the rookie mistake of releasing a lab result before it had been confirmed.
The old CDC would not have released surveillance reports and investigation outbreak reports containing multiple errors and/or inconsistencies.
This is what happens when the inmates are given the keys to the asylum’s executive offices.
It’s time to put control of these essential health agencies back into professional hands.
It’s time to change the locks.

Interested in learning more about food safety and the history of foodborne disease outbreaks and investigations?
Click on the link to listen to a short excerpt, then follow the buy links to add a digital, print or audio copy to your personal library.